H.S. Doris Mei and David F. Coker
Department of Chemistry
Boston University
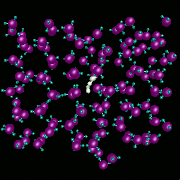
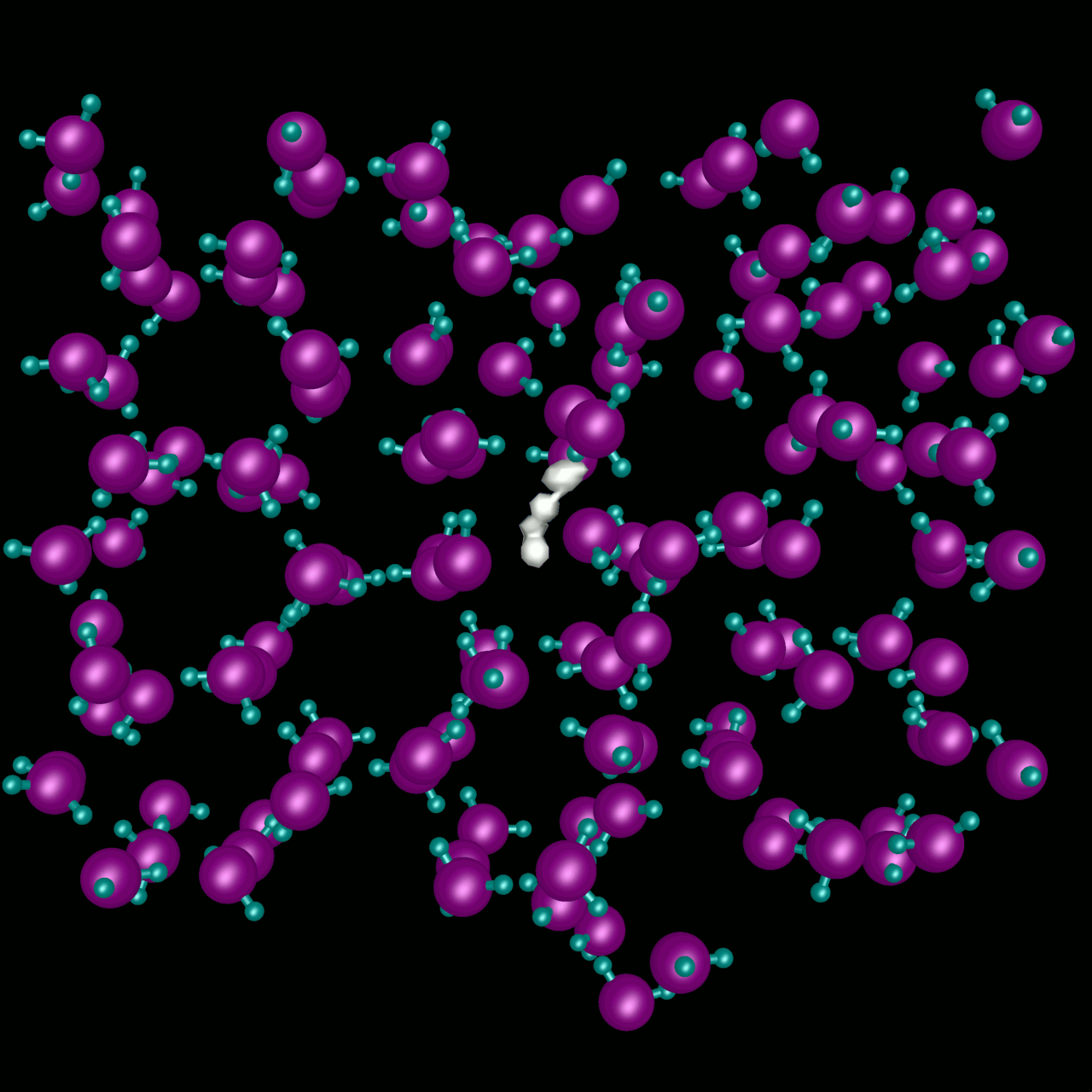
The transport of H2 in liquid water is studied using adiabatic, nonadiabatic, and classical molecular dynamics methods to understand the influence of transitions between translational states of the H2 molecule driven by solvent fluctuations. The video shows results from nonadiabatic dynamics calculations where we see the effects of these quantal transitions. Quantum autocorrelation functions of the H2 center-of-mass velocity are computed in various dynamical limits. We find strong nonadiabatic couplings between the instantaneous adiabatic translational states of H2 in water which result in rapid decorrelation of the H2 center-of-mass velocity for the time-evolving translational mixed state. Transitions to excited translational states dramatically reduce the effects of caging dynamics in the velocity autocorrelation function. Classical and adiabatic descriptions of the dynamics predict that caging is much more important than we find nonadiabatically. The diffusion constants obtained from these nonadiabatic calculations agree well with experimental results. Results obtained from other methods can be as much as a factor of two different from the experiment.
Video Sequences
{kind=link}
Non-adiabtic H2 in ice at T=250K
{kind=link}
Non-adiabtic H2 in water at T=300K
Hardware: Cray YMP and Convex C3880.
Software: Animations produced using the Boston University Modeling System visualization library and software packages.
Graphics programming and video production: Glenn Bresnahan, Scientific Computing and Visualization Group, Boston University.
Acknowledgments: We gratefully acknowledge financial support for this work from the National Science Foundation (Grant No. CHE-9058348 and CHE-9521793), and the Petroleum Research Fund administered by the American Chemical Society (Grant No. 27995-AC6), and a generous allocation of supercomputer time from the National Center for Supercomputing Applications.