Lintao Bu and John E. Straub
Department of Chemistry, Boston University
Introduction
At the most fundamental level, the process by which “biomolecular motors” convert chemical energy to heat and useful work must be understood in terms of molecular energy transfer and relaxation. We have employed atomic level models in dynamical simulations of vibrational energy transfer in proteins to (1) determine timescales for energy transfer, (2) isolate effective “pathways” for energy transfer, and (3) interpret experimental studies of ultrafast energy relaxation in heme proteins. Our results provide insight into the process by which proteins “funnel” energy to drive conformational changes and do useful work at the molecular level.
Why Vibrational Energy Relaxation ( VER )?
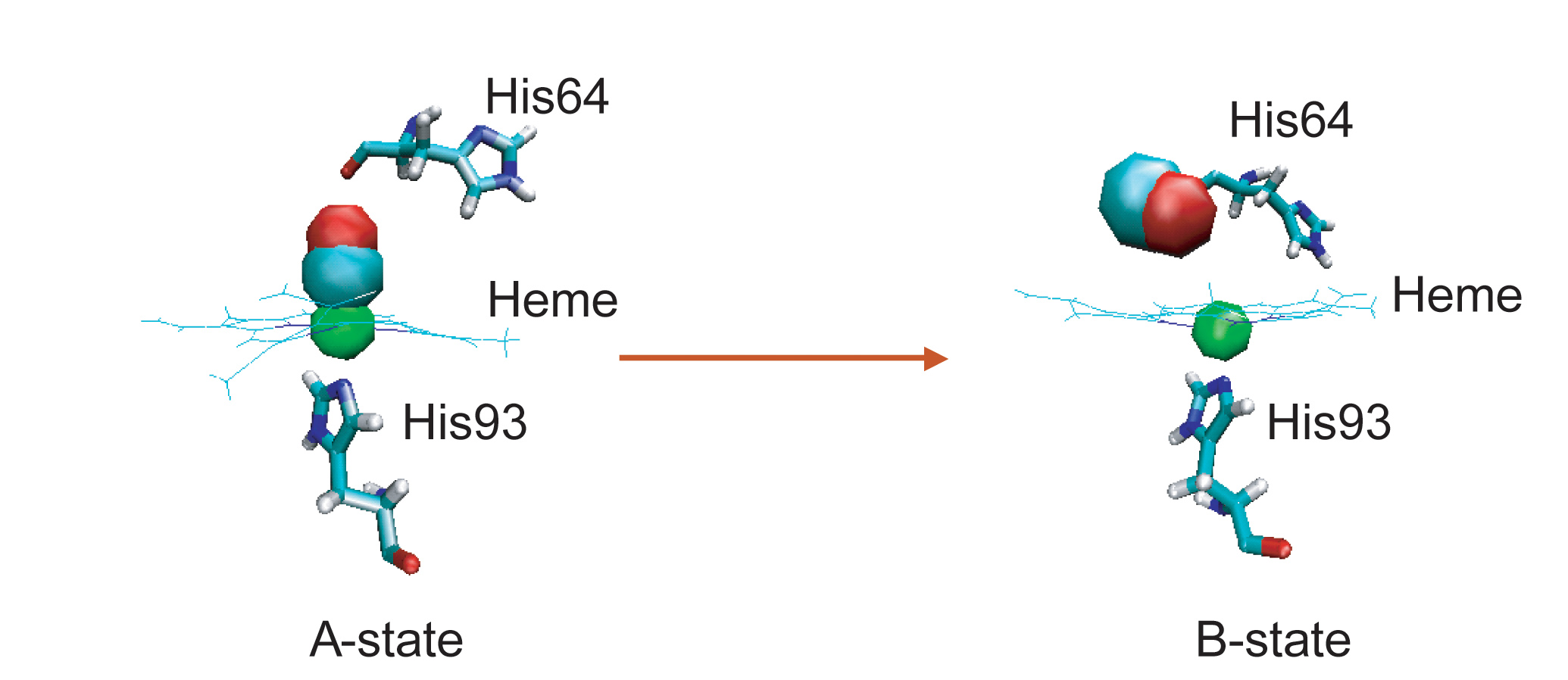
During VER, excess energy can flow into or out of reactive modes, thus controlling the rates of chemical reactions (ligand dissociation and rebinding in the protein).
Ligand dissociation can cause a global conformational transformation of the protein, providing a good system to study the cooperative nature of protein dynamics.
Simulation Model and Method

- Peptide + 2982 HO, 11499 atoms
- CHARMM 22 potential function
- 10 30ps MD trajectories at 300K
Computational Details
- The non-bonded potential was truncated using a group switching function extending from 9.5 to 11.5 A.
- SHAKE was employed to constrain bonds involving hydrogen atoms to their equilibrium values.
- A time step of integration of 1 fs was employed in the Verlet algorithm in the CHARMM program.
Is it possible that the vibrational energy of the heme could be transferred directly to the surrounding water by the solvent-induced damping of these stimulated [heme] side chains? There is no precedent for such highly directed energy funneling in molecular relaxation processes. – Hochstrasser
* Sagnella and Straub, J. Phys. Chem. 105, 7057 (2001). Lim, Jackson and Anfinrud, J. Phys. Chem. 100, 12043 (1996).
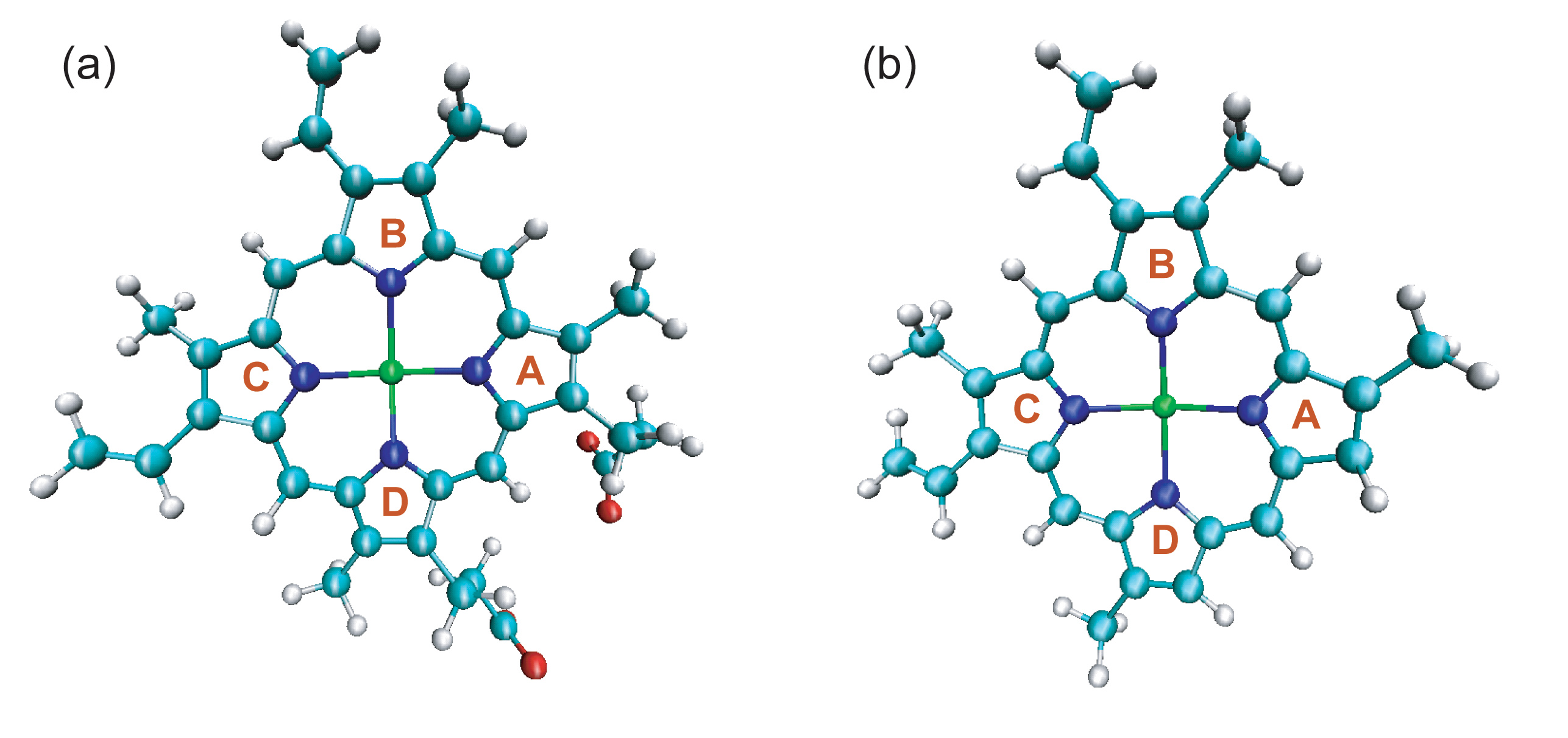
(a) The structure of heme for wild type/His93Gly mutant myoglobin, in which the positions of two propionate side chains can be seen.
(b) The structure of heme for the modified myoglobin, in which the two propionate side chains are amputated and replaced by two hydrogen atoms.

Hot protein residues in His93Gly mutant myoglobin and modified heme myoglobin depicted using space-filing CPK models. Red indicated the heme pocket region, green indicates the proximal His93/Gly93 residue.
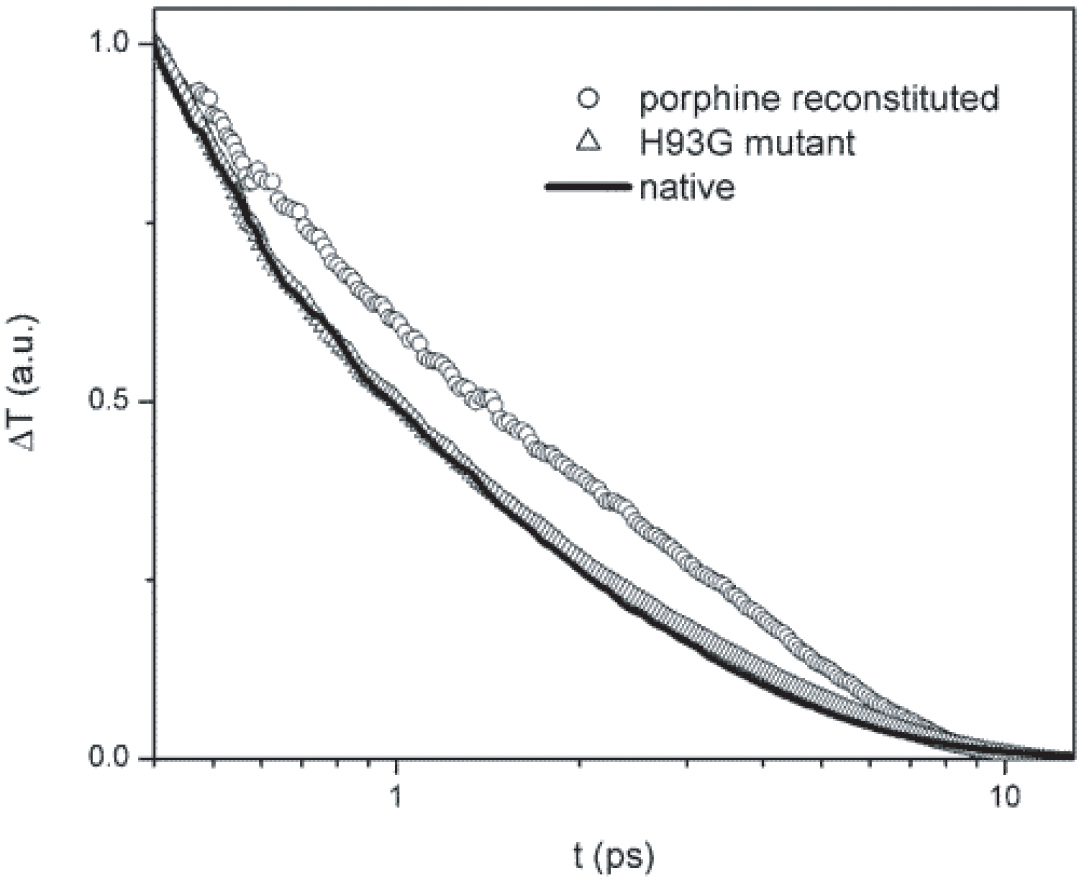
Optical transients of native Myoglobin, porphine-reconstituted Myoglobin, and H93G mutant Myoglobin. This experimental result is a strong support of our theoretical prediction. Vibrational relaxation path through the van der Waals contacts at the heme periphery is more important than through the single Fe-His covalent bond between the heme and the protein matrix. This experimental result is a strong support of our theoretical prediction.
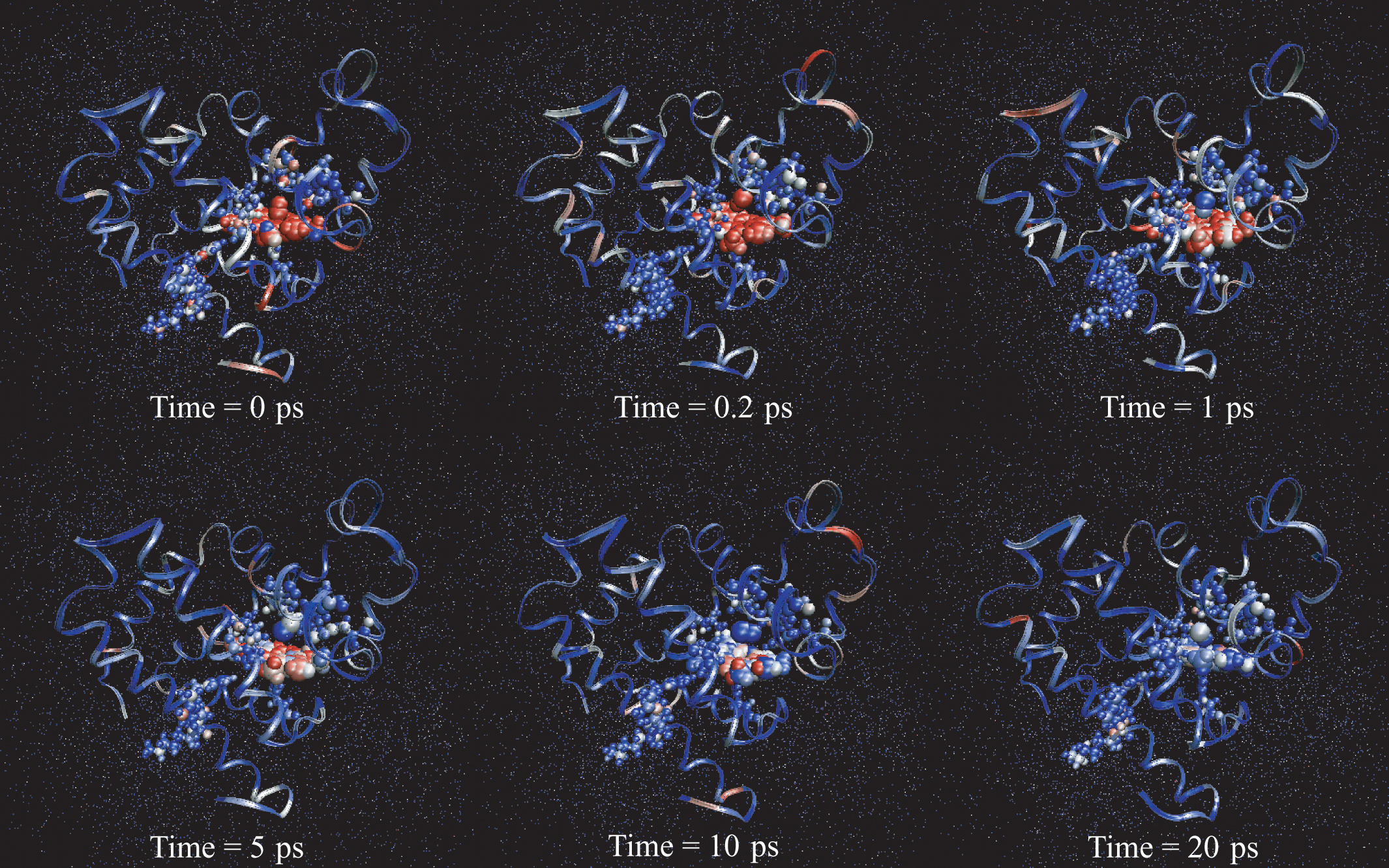
Redistribution of excess kinetic energy from the heme to the surrounding protein. Protein and heme atoms are shown as space-filling CPK modes. The solvent atoms are shown as dots. Each atom is colored on the basis of its temperature. Red corresponds to the regions of highest temperature, and blue marks the regions of lowest temperature.
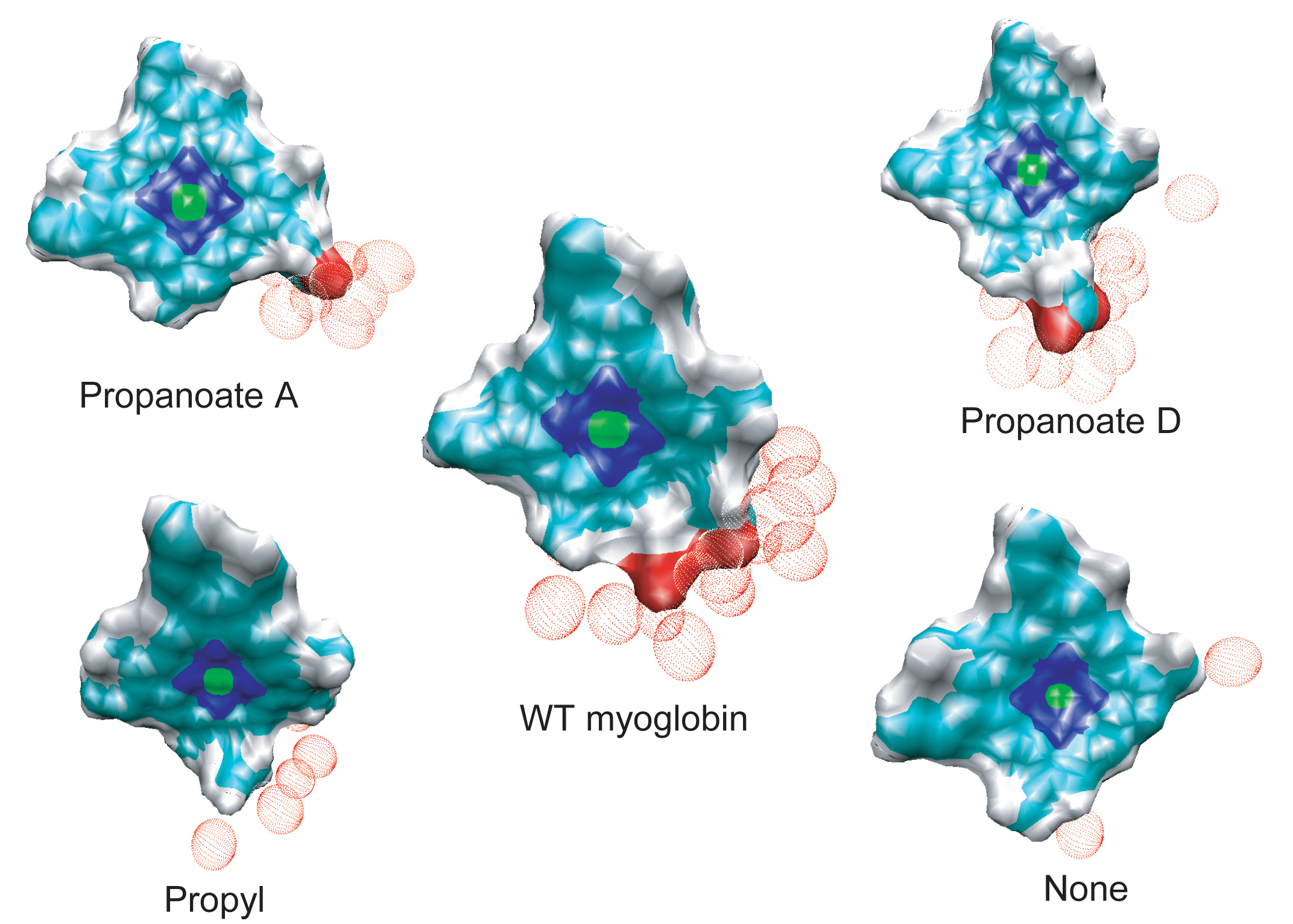
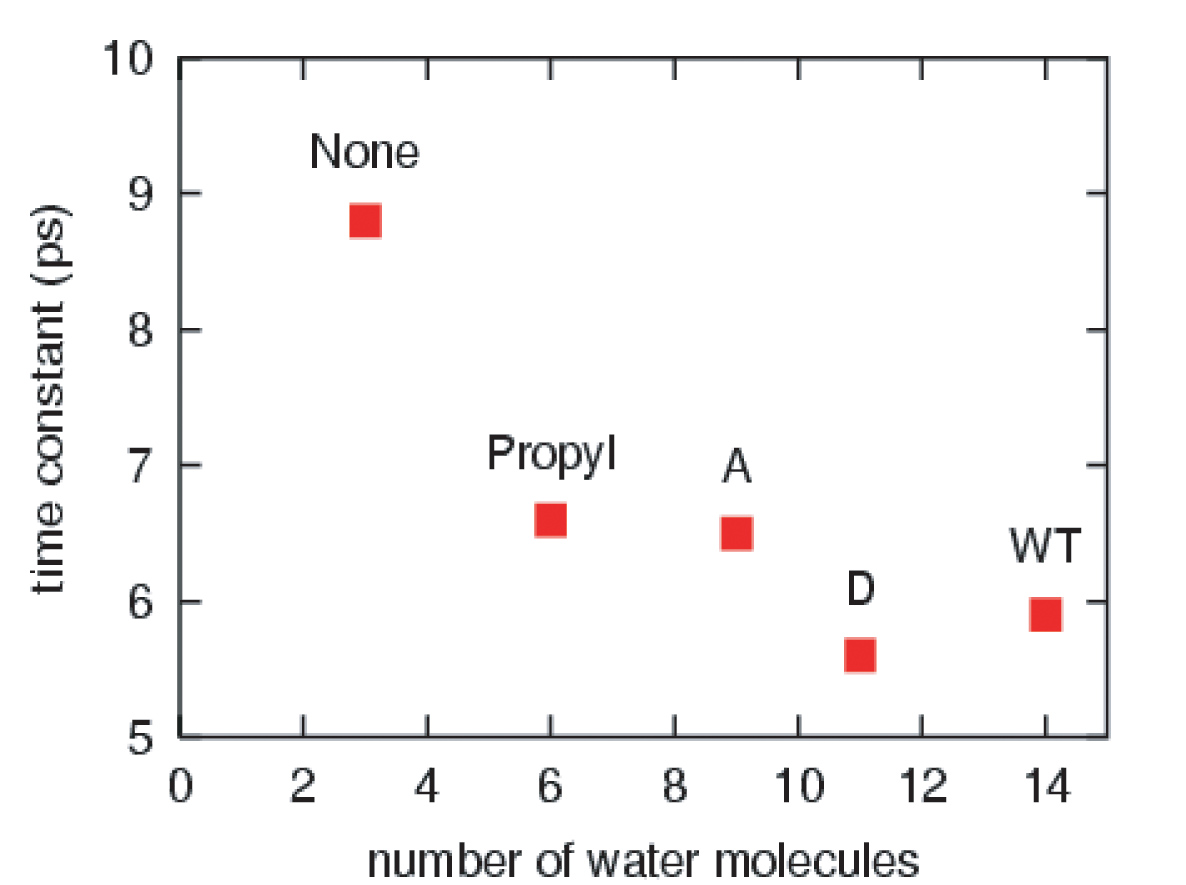
The effective solvation region around the heme plays the primary role in heme cooling. The hemes of wild type myoglobin and propanoate D mutant myoglobin have the largest effective solvation region, therefore they have he fastest cooling process. In decreasing the solvation region around the heme, the cooling rate of the hot heme will also decrease.
Summary
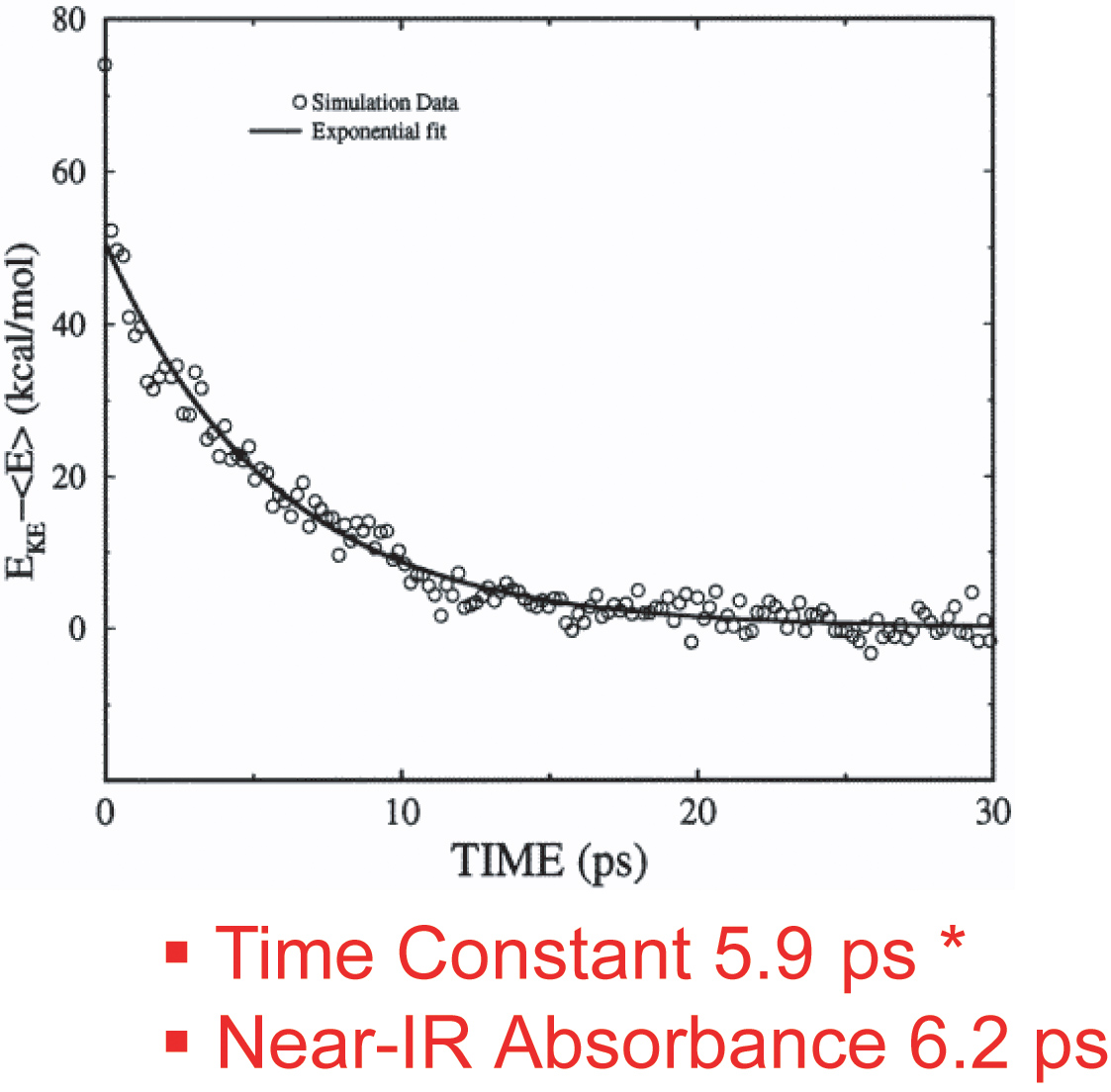
This work has investigated the time scales and pathways of vibrational energy relaxation in two modified forms of myoglobin following ligand photolysis. Our simulation results suggest that the excess kinetic energy of the heme dissipates according to a single exponential decay process with the relaxation time constant of 5.9 ps for the His93Gly mutant and 8.8 ps for the modified heme mutant. The rate of heme vibrational energy relaxation in wild type myoglobin with heme b was found to be 5.9 ps by simulation in agreement with the 6.2 ps time scale measured experimentally by Anfinrud and coworkers.
The cleavage of the single covalent bond between the heme and apoprotein in myoglobin does not cause any change in the rate or mechanism of vibrational energy relaxation of the heme. However, the amputation of the propionate side chains results in an increase in the vibrational energy relaxation time by 50%. There is a corresponding change in the mechanism of heme cooling with a significant pathway for energy flow being through space energy transfer from the heme to neighboring protein side chains. Our results support the conjecture that the two isopropionate side chains and their coupling to the solvent both play a dominant role in the dissipation of excess kinetic energy of the excited heme in solvated wild type myoglobin.
Acknowledgments
We are thankful for the generous support of this research by the National Science Foundation (CHE9975494 and CHE0316551). We are grateful to the Center for Computational Science and the Scientific Computing and Visualization group at Boston University for computational resources.