Targeting protein translation initiation for cancer therapy
Vajda, Porco, Kozakov, and Beglov groups, in collaboration with Jerry Pelletier (McGill University, Montreal)
Supported by:
NIH R01 GM064700 “Computational Mapping of Proteins for the Binding of Ligands” (PI: Vajda)
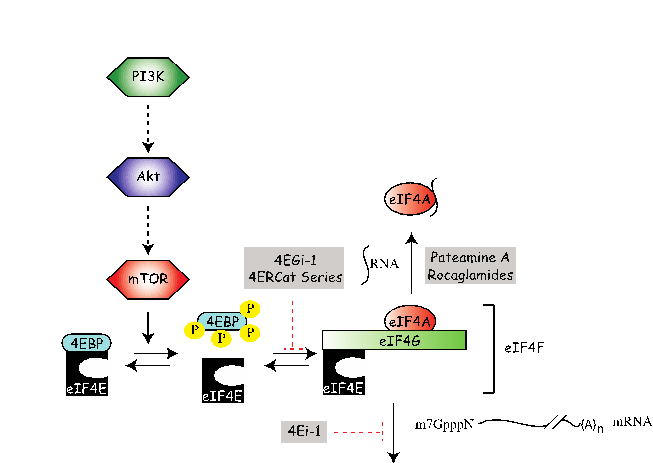
Protein synthesis presents an emerging class of cancer drug targets. The translation process occurs in three phases: initiation, elongation, and termination, with initiation being the rate-limiting. The ribosome recruitment phase of translation initiation is mediated by the eIF4 class of initiation factors (Fig. 1). The eIF4F complex, consisting of: (i) eIF4E, the cap-binding protein responsible for binding of eIF4F to the mRNA cap structure; (ii) eIF4A, an RNA helicase required to unwind local secondary structure and facilitate access of the 43S ribosomal complex to the mRNA template; and (iii) eIF4G, a scaffolding protein that mediates mRNA binding to the 43S pre-initiation complex, stimulates ribosome recruitment to mRNA templates. EIF4F complex assembly is controlled by the PI3K/Akt/mTOR signaling pathway, and this point of translational control is usurped in a majority of human cancers due to lesions in the PI3K/Akt signaling pathway. Given the large number of studies implicating eIF4F in supporting tumor initiation and maintenance, several labs have sought to identify small molecules that could be used to validate the anti-neoplastic potential of inhibiting eIF4F activity and possibly transformed into novel therapeutics.
One potential target to disrupt is the complex formed by eIF4E and eIF4G. The eIF4E-binding proteins (the best characterized one being 4E-BP1) compete with eIF4G for binding to eIF4E. Hypophosphorylated 4E-BP1 binds to eIF4E with high affinity (nanomolar range), whereas increased phosphorylation of 4E-BP1 by mTOR decreases the affinity for eIF4E, allowing eIF4E to assemble into the eIF4F complex with concomitant stimulation of translation [37]. The interaction between 4E-BP1 and eIF4E has been characterized by high resolution X-ray crystallography and NMR with many of the intermolecular contacts between these two proteins defined. 4EGI-1 from the Wagner lab (KD ~25 μM) inhibits cap-dependent translation, is toxic against several cancer cell lines [39], and is able to induce apoptosis in human lung cancer cells [40]. In collaboration with the Porco and Pelletier labs we have identified several inhibitors with IC50’s ranging from 4 to 40 M. All compounds block the interactions between eIF4E:eIF4G and between eIF4E:4E-BP1, as well as impair cap-dependent protein synthesis.
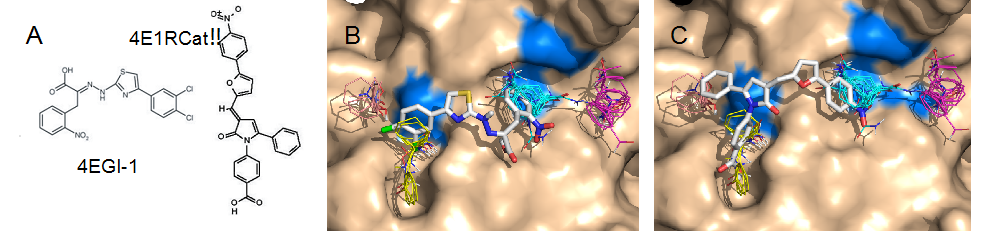
Our goal is to use novel computational approaches developed in the Vajda lab to craft these compounds into more potent and selective eIF4E:eIF4G inhibitors that will be synyhesized in the Porco lab, and tested in the lab of Dr. Pelletier at McGill University. In order to design new compounds, we first docked the known inhibitors to eIF4E. Fig. 2A shows the docking of inhibitors 4EGI-1 and 4E1RCat (the highest affinity inhibitor so far) to the eIF4E:eIF4G interaction site. Since 4EGI-1 binds to eIF4E but does not compete with mRNA binding, we blocked the mRNA cap (m7GpppN) binding site prior to mapping. Mapping free eIF4E showed four hot spots that form an elongated binding site (Figs. 2B&C). Although no eIF4E structure is available with bound inhibitor, line broadening observed by NMR indicates that residues H37, V69, L131, and I138 (colored blue in Fig. 2) on eIF4E interact with 4EGI-1. We used the correlation with probe density to find the docked pose of the inhibitors. Results show that the bound 4EGI-1 is in good agreement with the observed line broadening, but it occupies only two of the four hot spots (Fig. 2B) Fig. 2C shows that the slightly higher affinity inhibitor 4E1RCat occupies 3 of the 4 binding hot spots. We will build on these results, and will use density correlation (DC) as an additional term in the scoring function when selecting near-native poses among the ones generated by the docking. As shown in Figures 2B and 2C, none of these inhibitors reaches into the hot spot on the right, shown in magenta, and currently we focus on searching for compounds that will also utilize this region.
In a related research, the Porco group studied rocaglates/rocaglamides, a class of natural products known to display potent anticancer activity. They have shown that one such derivative, silvestrol, has activity comparable to taxol in certain settings. In addition, they reported the synthesis of various rocaglamide analogues and identification of a hydroxamate derivative (−)-9 having activity similar to silvestrol in vitro and ex vivo for inhibition of protein synthesis. It is assumed that silvestrol and the related analogues bind to eIF4A in the presence of mRNA, disrupting the 4A-4G interaction, but the location of binding is not known. We will use computational and NMR methods to determine the mode of the interaction, and possibly use this information to design better inhibitors of eIF4F action.