Hemoglobin Diagnostic Reference Laboratory
Director: Eric Burks, MD
Associate Director: Hong-yuan Luo, MB, PhD
 Laboratory Manager: Thomas Maher, MS
Laboratory Manager: Thomas Maher, MS
Hemoglobin Diagnostic Reference Laboratory
670 Albany Street, 3rd Floor Room 328
Boston MA 02118
Phone 617-414-5312
Fax: 617-414-5315
Email: hemoglobin@bmc.org
Billing Diagnostic Repertoire and Requisition Form
Globin Gene Mutations Diagnosed
|
|
|

- The α-globin gene cluster is on the short arm of chromosome 16.

- The β-globin gene cluster is on the short arm of chromosome 11.
- Globin gene mutations are the most common hereditary monogenic disease in man.
- There are now over 1,200 known natural globin gene mutations.
- These are tabulated in Globin Gene Server
- These mutations are found in ALL populations, but more prevalent in people from Africa, Mediterranean region, Eastern Europe, Middle East, Indian subcontinent, and southeast Asia, the so-called “malaria belt.”
- In some populations, carriers of sickle cell hemoglobin or thalassemia can range from 10% to 40%.
- With increasing racial and ethnic diversity in our country, hemoglobin disorders are now encountered more frequently than ever.
- With an increasing ethnic mix of populations, unusual combinations of globin gene mutations, each of which alone might be innocuous, could result in severe clinical syndromes.
- For the best possible patient care and counseling, accurate genetic diagnosis is required.

{kind=link}
Analyses
Our Laboratory specializes in hemoglobin and DNA-based mutational analyses to diagnose:
- Clinically important variant hemoglobins:
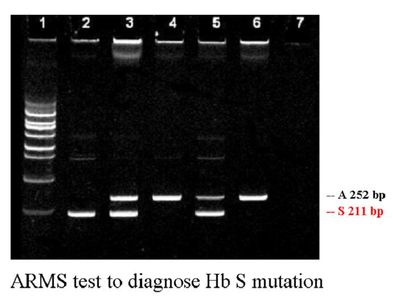
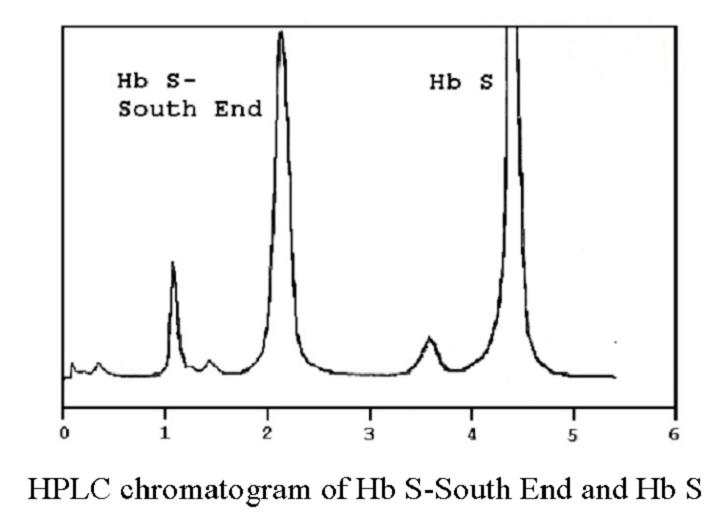
Sickle cell anemia, e.g. Hb S, C, D, O, Quebec-Chori, S-South End.
Hemolytic anemia caused by unstable variant hemoglobins.
Thalassemia, e.g. Hb E, Malay.
Erythrocytosis caused by high oxygen affinity variant hemoglobins.
Low blood oxygen saturation caused by low oxygen affinity variant hemoglobins.
Cyanosis caused by hereditary methemoglobinemias.
- Thalassemia mutations that markedly decrease or abolish globin chain production:
b–Thalassemias, both common and uncommon point mutations, and deletions.
a–Thalassemias, both deletions and point mutations.
- Hereditary persistence of fetal hemoglobin (HPFH)
Laboratory Correlation and Consultation
We provide clinical / genetic / laboratory correlation and consultation.
Blood Sample Required
- 2 tubes of EDTA-anticoagulated blood (lavender top tube), less in children.
- Please email hemoglobin@bmc.org for additional information or clarification.
Laboratory Certification
- Our laboratory is CAP certified (The College of American Pathologists)
- CLIA accredited (The Clinical Laboratory Improvement Amendments Center for Medicare and Medicaid Services
- Is an integral part of:
The Center of Excellence in Sickle Cell Disease
CPT Codes
- 83020 Hemoglobin fractionation and quantitation; electrophoresis
- 83021 Hemoglobin fractionation and quantitation; chromatography
- 81257 HBA1/HBA2 Common deletions or variants
- 81269 HBA1/HBA2 Gene Analysis DUP/DEL Variants
- 81259 HBA1/HBA2 Gene Analysis Full Gene Sequence
- 81363 HBB Duplication/Deletion Variants
- 81364 HBB Full Gene Sequence
- 81479 Unlisted Molecular Procedures
- G0452 Molecular Diagnostic Interpretation & Report