Improving the ClusPro Protein-Protein Docking Server
Sandor Vajda, Dima Kozakov, Ioannis Paschalidis, and Pirooz Vakili
Supported by:
NIH R35 GM118078 “Analysis and Prediction of Molecular Interactions” (PI: Sandor Vajda)
NSF DBI 1458509 “ABI Development: Utilization of diverse data in exploring protein‐protein interactions” (PI: Sandor Vajda)
NSF DBI 1759472 “Collaborative Research: ABI Development: The next stage in protein‐protein docking” (PI: Sandor Vajda)
Although this project is supported by funding only to the Vajda lab, Drs. Kozakov, Paschalidis, and Vakili have been collaborators on the work, and their funding supports related but more computationally and mathematically focused projects (see below). In fact, the automated protein-protein docking server ClusPro has been originally developed in the Vajda lab, but the other PIs made and continue making very important contributions to the methodology.
The current version 2.0 of ClusPro employs a three-stage procedure for protein-protein docking shown in Fig. 1. It starts with rigid body global search based on the Fast Fourier Transform (FFT) correlation approach that evaluates the energies of billions of docked conformations on a grid. ClusPro 2.0 uses PIPER, the first FFT based docking program that can work with pairwise interaction potentials. PIPER performs exhaustive evaluation of an energy function in discretized 6D space of mutual orientations of two proteins. 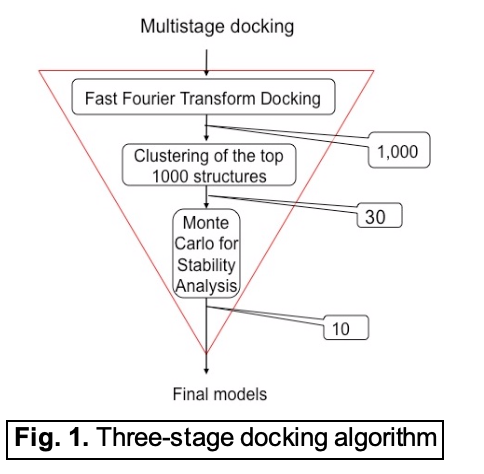 We sample 70,000 rotations, which approximately correspond to sampling at every 5 degrees in the space of Euler angles. In the translational space the sampling is defined by the 1.2 Å grid cell size, which means generating and evaluating up to 10 billion conformations for an average size complex. The complete scoring function of PIPER is given as the sum of terms representing shape complementarity, electrostatics, and chemical complementarity, the latter described by a pairwise interaction potential DARS [2]. The 1,000 lowest energy structures generated by PIPER are retained and clustered using pairwise Root Mean Square Deviation (RMSD) as the distance measure. The centers of the 30 largest clusters are the predicted structures of the complex, and are then refined by minimizing the Charmm energy of the docked models using a GBSA type solvation model.
We sample 70,000 rotations, which approximately correspond to sampling at every 5 degrees in the space of Euler angles. In the translational space the sampling is defined by the 1.2 Å grid cell size, which means generating and evaluating up to 10 billion conformations for an average size complex. The complete scoring function of PIPER is given as the sum of terms representing shape complementarity, electrostatics, and chemical complementarity, the latter described by a pairwise interaction potential DARS [2]. The 1,000 lowest energy structures generated by PIPER are retained and clustered using pairwise Root Mean Square Deviation (RMSD) as the distance measure. The centers of the 30 largest clusters are the predicted structures of the complex, and are then refined by minimizing the Charmm energy of the docked models using a GBSA type solvation model.
The main goal of the ongoing collaboration between the Vajda, Kozakov, Paschalidis, and Vakili groups is further improving the docking methods as follows.
Aim 1: Improved sampling with distance-dependent and multi-body scoring functions. Adding the structure-based pairwise contact potential DARS (Decoys as the Reference State) to the scoring function substantially improved the performance of ClusPro. However, distance-dependent and multi-body potentials showed definitive promise in the recent rounds of CAPRI. We are using the DARS approach to develop such scoring function terms. While similar potentials have been used by other groups for re-scoring docked structures, in ClusPro they will be used directly in the sampling stage. (Collaboration with the Kozakov group).
Aim 2: Improving docking and model selection by evolutionary considerations. Recently developed methods have shown considerable success in predicting residue–residue contacts in protein 3D interfaces using evolutionary covariance information. These methods require collecting evolutionary related sequences of interacting protein pairs and estimating residue-residue interactions. We will develop algorithms and software for estimating only inter-protein rather than intra-protein contacts. It is expected that restricting consideration to inter-protein pairs will enable reducing the number of sequences required for the estimation. The identified contacts will be used as distance restraints in ClusPro. (Collaboration with the Kozakov and Kon groups).
Aim 3: Exploring template-based docking and improving model selection and refinement by integration of direct and template-based methods. The main idea of template-based docking (TBD) is utilizing the known structures of protein complexes as templates to predict the quaternary structure of the target protein complex. Our goal is to implement TBD as part of the extended ClusPro server. Automating all steps of the algorithm, i.e., finding orthologs, aligning the appropriate sequences. We will consider modeling both homo and hetero-oligomers. The problems of optimally selecting either TBD or direct docking, and potentially combining the two approaches will also be considered.
Aim 4: Docking homology models. If template structures are available for the individual proteins but not for the complex, the server will need to perform direct docking of homology models that may include poorly resolved regions such as flexible loops. Direct focused docking can also be beneficial if the available templates are not very good due to limited sequence similarity, or due to structural differences among templates that are not distinguishable based on sequence identity.
Aim 5: Docking proteins with backbone conformational change upon binding. The problem is similar to the docking of homology models, since flexible regions of the structures, including loops and short linear motifs (SLIMs) connecting globular parts, generally have no well-defined conformations. Therefore, we decompose the proteins into rigid and flexible regions, and generate an ensemble of structures, first docking only the rigid parts.
Aim 6: Implementation of the novel algorithms in the ClusPro 3.0 server. The programs will be developed using current software engineering practices, and will be added as extensions to the web accessible public server ClusPro. The server will be implemented on a number of platforms, including computer clusters and multi-core desktops. All software produced will be free for academic and governmental use.
Publications:
Zarbafian S, Moghadasi M, Roshandelpoor A, Nan F, Li K, Vakli P, Vajda S, Kozakov D, Paschalidis IC. Protein docking refinement by convex underestimation in the low-dimensional subspace of encounter complexes. Sci Rep. 2018 Apr 12;8(1):5896. doi: 10.1038/s41598-018-23982-3.
Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein-protein docking. Nat Protoc. 2017 Feb;12(2):255-278. doi: 10.1038/nprot.2016.169. Epub 2017 Jan 12.
Padhorny D, Kazennov A, Zerbe BS, Porter KA, Xia B, Mottarella SE, Kholodov Y, Ritchie DW, Vajda S, Kozakov D. Protein-protein docking by fast generalized Fourier transforms on 5D rotational manifolds. Proc Natl Acad Sci U S A. 2016 Jul 26;113(30):E4286-93. doi: 10.1073/pnas.1603929113.
Mamonov AB, Moghadasi M, Mirzaei H, Zarbafian S, Grove LE, Bohnuud T, Vakili P, Paschalidis IC, Vajda S, Kozakov D. Focused grid-based resampling for protein docking and mapping. J Comput Chem. 2016 Apr 30;37(11):961-70. doi: 10.1002/jcc.24273.
Moghadasi M, Mirzaei H, Mamonov A, Vakili P, Vajda S, Paschalidis ICh, Kozakov D. The impact of side-chain packing on protein docking refinement. J Chem Inf Model. 2015 Apr 27;55(4):872-81. doi: 10.1021/ci500380a.
Chowdhury R, Beglov D, Moghadasi M, Paschalidis IC, Vakili P, Vajda S, Bajaj C, Kozakov D. Efficient Maintenance and Update of Nonbonded Lists in Macromolecular Simulations. J J Chem Theory Comput. 2014 Oct 14;10(10):4449-4454.